New & Noteworthy
Introducing BLAST Search for SGD at the Alliance of Genome Resources
February 17, 2026

We’re excited to announce the launch of a new BLAST service for Saccharomyces Genome Database (SGD) data, now available at the Alliance of Genome Resources. This release marks another significant milestone in our ongoing effort to migrate SGD services and data to the Alliance platform, ensuring continued access to essential yeast genomics tools within an integrated, multi-organism framework.
What’s New
The Alliance BLAST service provides researchers with powerful sequence similarity search capabilities against SGD datasets, maintaining the functionality that the yeast research community has relied on for years while benefiting from the Alliance’s modern infrastructure and cross-species integration.
Key Features
- Comprehensive SGD datasets: Search against the complete S. cerevisiae genome, including coding sequences, proteins, and genomic DNA, or search against genomes from dozens of other strains
- Familiar BLAST functionality: All standard BLAST algorithms (BLASTN, BLASTP, BLASTX, TBLASTN, TBLASTX) are supported
- Enhanced performance: Leveraging the Alliance’s updated infrastructure for faster search results
- Cross-species exploration: Seamlessly explore homologs across other Alliance member databases
Part of a Broader Migration
This BLAST service is part of our comprehensive strategy to transition SGD resources to the Alliance of Genome Resources. This migration ensures that:
- SGD data remains accessible and well-maintained for the research community
- Users benefit from integration with data from other model organisms
- Resources are consolidated on a sustainable, collaborative platform
- The yeast community gains access to enhanced comparative genomics tools
Getting Started
Visit the Alliance of Genome Resources to access the new service for SGD BLAST, including Fungal BLAST. The datasets will be familiar to longtime SGD users.
Looking Ahead
We remain committed to supporting the yeast research community through this transition. Additional SGD tools and features will continue to migrate to the Alliance platform in the coming months. Stay tuned for updates, and as always, we welcome your feedback.
Categories: Announcements
SGD Newsletter, December 2025
December 11, 2025
About this newsletter:
This is the December 2025 issue of the SGD newsletter. The goal of this newsletter is to inform our users about new features in SGD and to foster communication within the yeast community.
Contents
- 1 Support SGD this Holiday Season
- 2 New at SGD: Links to a Comprehensive Overview of Yeast Libraries
- 3 Retirement of Mike Cherry
- 4 Reminder: Submit Data Form
- 5 Alliance of Genome Resources News
- 6 microPublications – Latest Yeast Papers
- 7 Upcoming Conferences & Courses
- 8 Happy Holidays from SGD!
Support SGD this Holiday Season

Budget cuts from NIH continue to strain SGD’s finances. Despite our efforts at reducing costs, we still have significant ongoing budgetary challenges. Donations are now critical for our work to continue. Your generous gift to SGD enables us to continue providing essential information for your research and teaching efforts. We are now able to accept gifts via credit card. To contribute using a credit card, please use this form: give.stanford.edu.
- Under ‘Direct your gift,’ select ‘Other Stanford Designation’ from the pulldown menu
- In the ‘Other’ text box, specify SGD by including the text “Saccharomyces Genome Database – Account : GHJKO, Genetics : WAZC”
- Complete the form and payment to donate to SGD
If you’d like to contribute by check, please contact us at: sgd-helpdesk@lists.stanford.edu
We thank you in advance for your support!
New at SGD: Links to a Comprehensive Overview of Yeast Libraries

Baruch et al. have compiled and published A comprehensive overview of yeast libraries and their role in advancing cell biology, which describes a comprehensive list of genome-wide collections of Saccharomyces cerevisiae. They have grouped similar strain libraries together and have curated detailed descriptions, mating types, genotypes, references, and genome coverage. This information can help researchers identify strain libraries of interest to them. All of this information can also be found in tables on LibrarYeast. We have added links to these tables under the Resources section of individual Protein, Phenotype, and Interaction tabs.
Retirement of Mike Cherry

This year, we celebrate and thank J. Michael “Mike” Cherry as he retires after decades of visionary leadership of the Saccharomyces Genome Database. From the earliest days of the yeast genome to today’s multi‑omic era, Mike helped shape SGD into the trusted, rigorously curated resource at the heart of the yeast community. His commitment to high‑quality data, interoperability, and open science empowered discoveries around the world, and his mentorship nurtured generations of curators, developers, and collaborators. Mike’s steady guidance, big‑picture thinking, and relentless focus on serving researchers have left a lasting legacy that will continue to guide SGD’s mission.
On November 14th, many past and current Cherry lab members gathered at Stanford to celebrate Mike’s career. Thank you to everyone who attended, and especially to everyone that shared stories or memories from the past 3 decades of SGD. Gavin Sherlock is SGD’s new PI, and he is hardly a stranger to SGD! Mike and Gavin published their first article together about SGD in Science all the way back in 1998: Chervitz et al., Comparison of the Complete Protein Sets of Worm and Yeast: Orthology and Divergence.
On behalf of the SGD team and the global yeast community, we extend our deepest gratitude to Mike and wish him joy and fulfillment in the next chapter.
Thank you, Mike, for everything.

Reminder: Submit Data Form
If you’re itching for something to do over the winter break, have you checked your newly published data at SGD lately? SGD can use your help! Authors can submit data and information about their publications by pointing us to novel results, datasets (we appreciate GEO accession IDs!), or other important information, using SGD’s simple “Submit Data” form: https://www.yeastgenome.org/submitData.
Alliance of Genome Resources News
Alliance of Genome Resources – Latest Release 8.2.0
The 8.2.0 release includes data refreshes from each of the model organism source databases as well as various backend improvements. The sequence viewers on the gene and allele pages have been restored on gene pages, in the Alleles and Variants and Sequence Feature Viewer sections, and on allele pages, in the Genomic Variant Information section (see for example the mouse gene Pten and mouse allele for Pten). Also, on the gene page Alleles and Variants section, interactions between the variant sequence viewer and the alleles/variants table have been restored. Website infrastructure is upgraded to React 19 and migrated to Vite.
Alliance Webinars: MicroPublication Biology
On November 20, Tim Schedl, microPublication Biology Editorial Board member, presented an overview of the MicroPublication journal which publishes brief scholarly reports of research findings based on data presented in a single figure. View the recording of the latest Alliance of Genome Resources webinar: Introduction to MicroPublication Biology. Webinars are also planned for 2026, in January, February, and March. See the Event Calendar for the schedule of upcoming Alliance office hours and webinars.
FlyBase End of Funding
The termination of the NIH/NHGRI FlyBase grant has placed the long-term sustainability of FlyBase at risk. However, thanks to the generous support of several key individuals and institutions, FlyBase has announced that they will remain operational through the coming year. Looking ahead, ensuring FlyBase’s sustainability beyond the next year – and successfully integrating with the Alliance – will require new funding sources. FlyBase, like SGD, is asking for continued donations from any and all Fly supporters. For more information on how to support FlyBase, see the FlyBase wiki:
- European labs: Please consider contributing to the Cambridge, U.K. FlyBase group
- U.S. and other non-European labs: Please consider contributing to the U.S. FlyBase groups
Final Release of WormBase
WormBase announced their final release, WS298, on November 27, 2025. The existing WormBase website will continue provide access to this archived WS298 release of WormBase, and the content hosted there will no longer be updated except for essential maintenance to ensure continued availability. Ongoing curation, annotation, data updates, and new data for Caenorhabditis elegans will now be managed through the Alliance of Genome Resources. Users should refer to the Alliance for the most current worm datasets and continued resource development. Read more on the WormBase blog.
microPublications – Latest Yeast Papers

microPublication Biology is part of the emerging genre of rapidly-published research communications. microPublications publishes brief, novel findings, negative and/or reproduced results, and results which may initially lack a broader scientific narrative. Each article is peer-reviewed, assigned a DOI, and indexed through PubMed and PubMedCentral. Consider microPubublications when you have a result that doesn’t necessarily fit into a larger story, but will be of value to others. Latest yeast microPublications:
- King A, Emery TM, Reese K, Tinsley HN (2025) Yeast gene of unknown function YGL081W is involved in DNA damage response. MicroPubl Biol 2025
- Avogo EW, Burlingame NA, Badenahalli Narasimhaiah S, Delorme-Axford E (2025) Bioinformatics analysis identifies Mot2 protein as a potential regulator of autophagy in Saccharomyces cerevisiae. MicroPubl Biol 2025
- Nnabuenyi N, Sands MA, Camlin NJ (2025) Specific auxin and medium combinations alter Saccharomyces cerevisiae growth. MicroPubl Biol 2025
- Sasahara M, Matsumoto S, Tamura Y (2025) Dnm1 Is Required for the Focal Clustering of Fis1 on the Mitochondrial Outer Membrane. MicroPubl Biol 2025
- Dutca LM, Freed EF, Baserga SJ (2025) Yeast pre-rRNA is processed at the A’ site. MicroPubl Biol 2025
- Schiemann AH, Sarwar M, Sattlegger E (2025) Functional Analysis of the Yeast Counterpart to the Human GCN2 p.Glu738_Asp739insArgArg Variant. MicroPubl Biol 2025
- Torvi JR, Wong J, Drubin DG, Barnes G (2025) Stu2 is required for plus-end directed chromosome transport along microtubules during metaphase in Saccharomyces cerevisiae. MicroPubl Biol 2025
- VanderVen K, Butcher C, Fokine R, Li J (2025) Pep12 is important for proteasome microautophagy under low glucose conditions. MicroPubl Biol 2025
- Tang L, AlKaabi A, Meyer D (2025) Copper Homeostasis is influenced by Ics3 in Saccharomyces cerevisiae. MicroPubl Biol 2025
- Taguchi S, Matsuzawa R, Suda Y, Irie K, Ozaki H (2025) Investigating the effects of liquid handling robot pipetting speed on yeast growth and gene expression using growth assays and RNA-seq. MicroPubl Biol 2025
- Takesue H, Okada S, Ito T (2025) Long-read plasmid sequencing strategy for evaluating intrinsic instability of tandem gene arrays. MicroPubl Biol 2025
Upcoming Conferences & Courses
- 33rd Fungal Genetics Conference
- March 17 to March 22, 2026 –
- Asilomar Conference Grounds, Pacific Grove, CA
- Yeast Genetics Meeting 2026
- June 13 to June 17, 2026 –
- Asilomar Conference Grounds, Pacific Grove, CA
- 39th International Specialized Symposium on Yeasts (ISSY39)
- Nov 8 – 12, 2026
- Jeongdong 1928 Art Center, Seoul, KR
Happy Holidays from SGD!

We want to take this opportunity to wish you and your family, friends, and lab mates the best during the upcoming holidays. Stanford University will be closed for two weeks starting end of day Friday, December 19th, and reopening on Monday, January 5th, 2026. Although SGD staff members will be taking time off, the website will be up and running throughout the winter break, and we will resume responding to user requests and questions in the new year.
Note: If you wish to receive this newsletter via email, please contact the SGD Help Desk at sgd-helpdesk@lists.stanford.edu.
Categories: Newsletter
SGD Newsletter, Spring 2025
April 30, 2025
About this newsletter:
This is the Spring 2025 issue of the SGD newsletter. The goal of this newsletter is to inform our users about new features in SGD and to foster communication within the yeast community.
Contents
- Give a Gift / Support SGD: Credit Cards Now Accepted
- SGD’s Latest Genetics Publication
- Pathway Annotations Now Available as GO Annotations
- New Yeast Phenome Links in Phenotype Resources
- microPublications – Latest Yeast Papers
- Alliance of Genome Resources – Latest Release 8.1.0
- Upcoming Conferences & Courses
Give a Gift / Support SGD: Credit Cards Now Accepted
Budget cuts from NIH continue to strain SGD’s finances. Despite our efforts at reducing costs, we still have significant ongoing budgetary challenges. Donations are now critical for our work to continue.
Your generous gift to SGD enables us to continue providing essential information for your research and teaching efforts. We are now able to accept gifts via credit card.
To contribute using a credit card, please use this form: give.stanford.edu.
- Under ‘Direct your gift,’ select ‘Other Stanford Designation’ from the pulldown menu
- In the ‘Other’ text box, specify SGD by including the text “Saccharomyces Genome Database – Account : GHJKO, Genetics : WAZC”
- Complete the form and payment to donate to SGD
If you’d like to contribute by check, please contact us at: sgd-helpdesk@lists.stanford.edu
Thank you for your support!
SGD’s Latest Genetics Publication

Saccharomyces Genome Database: Advances in Genome Annotation, Expanded Biochemical Pathways, and Other Key Enhancements has now been published in GENETICS and is available in the March 2025 issue.
Check out the most recent updates at SGD, including:
- Two most recent reference genome annotation updates
- Expanded biochemical pathways representation
- Changes to SGD search and data files
- Other enhancements to the SGD website and user interface
Pathway Annotations Now Available as GO Annotations

YeastPathways, the database of metabolic pathways and enzymes in the budding yeast Saccharomyces cerevisiae, is manually curated and maintained by the biocuration team at SGD.
This resource is jam-packed with information, but was somewhat hidden from view. We have been doing different things recently to make the pathways more readily accessible. Initially, we added a new section with pathways links on the relevant gene pages (ex. DFR1). Additionally, we made the pathways available in SGD Search. Now, we have transformed the metabolic pathways and associated genes/enzymes into Gene Ontology (GO) annotations (ex. DFR1).
Because many fundamental molecular processes and pathways are evolutionarily conserved between yeast and higher eukaryotes, including humans, the curated metabolic pathway information has great value for the transfer of knowledge to other organisms. It is for this reason that the YeastPathways data were exported in BioPAX (Demir et al. 2010) format for import into Noctua, a tool for collaborative curation of biological pathways and gene annotations that was developed by the GO Consortium (Thomas et al. 2019). BioPAX provides a standardized format for representing biological pathways, allowing researchers to integrate pathway information from different sources and databases. Noctua can import pathway data encoded in BioPAX format to populate the pathway editor with molecular interactions, biological processes, and regulatory relationships, and can utilize BioPAX files to combine pathway data from multiple datasets for pathway curation and analysis.
Pathways curated and edited in Noctua can be exported both as GO annotations for yeast and orthologous genes in other species, or as pathway annotations in BioPAX, facilitating the sharing of curated pathways with other researchers, databases, and analysis tools using a standard format, promoting data exchange, and collaboration within the scientific community.
New Yeast Phenome Links in Phenotype Resources
The Yeast Phenome is a collaborative project from the Baryshnikova lab at Calico Life Sciences to create a comprehensive compendium of systematic loss-of-function phenotypes for the budding yeast Saccharomyces cerevisiae.
The Yeast Phenome systematically tracks, collects, and annotates all published phenotypic screens utilizing the yeast knock-out collection.
Locus-specific links to the Yeast Phenome are now available in the Resources section of the Phenotype tab and will take you directly to the corresponding page of the Yeast Phenome data library. Try it now: the SGD phenotype page for ARP1, and the linked ARP1 page at Yeast Phenome.
microPublications – Latest Yeast Papers
microPublication Biology is part of the emerging genre of rapidly-published research communications. microPublications publishes brief, novel findings, negative and/or reproduced results, and results which may initially lack a broader scientific narrative. Each article is peer-reviewed, assigned a DOI, and indexed through PubMed and PubMedCentral.
Consider microPubublications when you have a result that doesn’t necessarily fit into a larger story, but will be of value to others.
Latest yeast microPublications:
- Andrade Latino A, Biggins S (2025) Analysis of a cancer-associated mutation in the budding yeast Nuf2 kinetochore protein. MicroPubl Biol 2025
- Butcher C, VanderVen K, Li J (2025) Proteasome condensates repeatedly “contact and release” at the nuclear periphery during dissolution. MicroPubl Biol 2025
- Eftimie A, Meyer D (2025) Transcription Regulatory Protein SIN3 (YOL004W) Influences Mutation Rates in Saccharomyces cerevisiae. MicroPubl Biol 2025
- James M, Klain GK, Brito SO, Trejo L, Okello TMA, Segarra VA (2025) Autophagy-deficient budding yeast cells are sensitive to freeze-thaw stress. MicroPubl Biol 2025
- Law S, et al. (2024) The Role of hBRCA2 in the Repair of Spontaneous and UV DNA Damage in Saccharomyces cerevisiae. MicroPubl Biol 2024
- Pfliegler WP, Imre A, Biotechnology BSc Class Of UOD, Pócsi I (2025) PCR-fingerprinting of culturable yeasts from commercially obtained beers: a simple and engaging applied microbiological laboratory exercise. MicroPubl Biol 2025
- Thota K, Fredette-Roman JD, Sharp NP (2025) Yeast mutation rates in alternative carbon sources reflect the influence of reactive oxygen species. MicroPubl Biol 2025
All yeast microPublications can be found in SGD.
Alliance of Genome Resources – Latest Release 8.1.0
The 8.1.0 release includes data refreshes from each of the model organism source databases as well as various backend improvements.
Recent changes in 8.0.0 and 8.1.0 include:
- Redesign of the legend corrects issues with casual relationship representation; interface visuals have been refined; all defined causal relationships are now depicted using standardized glyphs with a matching color legend.
There is now an Event Calendar with the schedule of upcoming Alliance office hours and webinars: https://www.alliancegenome.org/event-calendar
- View the video of the latest Alliance of Genome Resources webinar:
- How AI informs literature curation at the Alliance
- Presented on April 17, 2025 by Kimberly Van Auken and Valerio Arnaboldi of WormBase, and Chris Tabone of FlyBase.
Upcoming Conferences & Courses
- Gene Ontology Consortium Spring 2025 Meeting
- May 5-8, 2025
- Geneva, Switzerland
- SGD will be attending
- CSH Asia: Yeast and Life Sciences
- June 02 to June 06, 2025 –
- Suzhou, China
- PNWYC 2025: Pacific Northwest Yeast Club
- June 20, 2025
- Vancouver, Canada
- Yeast2025: 32nd International Conference on Yeast Genetics and Molecular Biology ICYGMB32
- July 21 to July 24, 2025
- Sorbonne University, Paris, France
- SGD will be attending
- Yeast Genetics & Genomics
- July 22 to August 12, 2025
- Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
- SGD will be attending
- 38th International Specialized Symposium on Yeasts (ISSY38)
- September 01 to September 05, 2025 –
- Warsaw University, Warsaw, Poland
- 33rd Fungal Genetics Conference
- March 17 to March 22, 2026 –
- Asilomar Conference Grounds, Pacific Grove, CA
Categories: Newsletter
Yeast Biochemical Pathways incorporated into Gene Ontology annotations
April 09, 2025
YeastPathways, the database of metabolic pathways and enzymes in the budding yeast Saccharomyces cerevisiae, is manually curated and maintained by the biocuration team at SGD.
This resource is jam-packed with information, but somewhat hidden from view. We have been doing different things recently to make the pathways more readily accessible. Some time ago we added a new section with pathways links on the relevant gene pages (ex. DFR1).

We also made the pathways available in SGD Search.

Now we have transformed the metabolic pathways and associated genes/enzymes into Gene Ontology (GO) annotations (ex. DFR1).

Because many fundamental molecular processes and pathways are evolutionarily conserved between yeast and higher eukaryotes, including humans, the curated metabolic pathway information has great value for the transfer of knowledge to other organisms. It is for this reason that the YeastPathways data were exported in BioPAX (Demir et al. 2010) format for import into Noctua, a tool for collaborative curation of biological pathways and gene annotations that was developed by the GO Consortium (Thomas et al. 2019). BioPAX provides a standardized format for representing biological pathways, allowing researchers to integrate pathway information from different sources and databases. Noctua can import pathway data encoded in BioPAX format to populate the pathway editor with molecular interactions, biological processes, and regulatory relationships, and can utilize BioPAX files to combine pathway data from multiple datasets for pathway curation and analysis.
Pathways curated and edited in Noctua can be exported both as GO annotations for yeast and orthologous genes in other species, or as pathway annotations in BioPAX, which facilitates the sharing of curated pathways with other researchers, databases, and pathway analysis tools using a standard format, promoting data exchange, and collaboration within the scientific community.
Categories: Data updates
Apply Now for the 2025 Yeast Genetics and Genomics Course
March 11, 2025

For over 50 years, the legendary Yeast Genetics & Genomics course has been taught each summer at Cold Spring Harbor Laboratory, though the name didn’t include “Genomics” in the beginning. The list of people who have taken the course reads like a Who’s Who of yeast research, including Nobel laureates and many of today’s leading scientists.
The application deadline is April 15, so don’t miss your chance!
Find all the details and application form at the CSHL Meetings & Courses site. This year’s instructors – Grant Brown, Soni Lacefield, and Greg Lang – have designed a course (July 22 – August 12) that provides a comprehensive education in all things yeast, from classical genetics through up-to-the-minute genomics. Students will perform and interpret experiments, learning about things like:
- Transformation & Genome Engineering
- Microscopy
- Manipulating Yeast
- Dissecting Tetrads
- Isolating Mutants
- Working with Essential Genes
- Synthetic Genetic Arrays
- Fluctuation Assays
- Whole Genome Sequencing & Analysis
2025 Invited Speakers
Francisco Cubillos, Universidad de Santiago de Chile, Chile
Ivan Dedek, Meier’s Creek Brewing Company, Cazenovia, NY
Ian Ehrenreich, University of Southern California, Los Angeles, CA
Catherine Freudenreich, Tufts University, Medford, CT
Jonathan Friedman, University of Texas Southwestern, TX
David Garcia, University of Oregon, Eugene, OR
Kerry Geiler-Samerotte, Arizona State University, Tempe, AZ
Patrick Gibney, Cornell University, Ithaca, NY
Elena Kuzmin, Concordia University, Montreal, Canada
Jun-Yi Leu, Academia Sinica, Taipei, Taiwan
Amy MacQueen, Wesleyan University, Middletown, CT
Rob Nash, Saccharomyces Genome Database, Stanford University, Palo Alto, CA
Ally O’Donnell, University of Pittsburgh, Pittsburgh, PA
Techniques have been summarized in the accompanying course manual, published by CSHL Press.

Who should attend? Scientists who aren’t part of large, well-known yeast labs are especially encouraged to apply – for example, professors and instructors who want to incorporate yeast into their undergraduate genetics classrooms; scientists who want to transition from mathematical, computational, or engineering disciplines into bench science; and researchers from small labs or institutions where it would otherwise be difficult to learn the fundamentals of yeast genetics and genomics.
What else goes on there? Besides its scientific content, the fun and camaraderie at the course is also legendary. In between all the hard work there are late-night chats at the bar and swimming at the beach. There’s a fierce competition between students at the various CSHL courses in the Plate Race, which is a relay in which teams have to carry stacks of 40 Petri dishes (used, of course). There’s also typically a sailboat trip, a microscopy contest, and a mysterious “Dr. Evil” lab!
The Yeast Genetics & Genomics Course is loads of fun – don’t miss out!
Categories: Conferences
Give a Gift / Support SGD
January 31, 2025
Giving to SGD just got easier! We now accept donations by credit card with this form: give.stanford.edu.
Select ‘Other Stanford Designation’ under ‘Direct your gift’ & in the ‘Other’ box, add: Saccharomyces Genome Database – Account: GHJKO, Genetics: WAZC. Thanks for your support!
Your generous gift to SGD enables us to continue providing essential information for your research and teaching efforts. Donations are now critical for our work to continue.
To contribute via check, please make checks payable to Stanford University, and include a note stating that “these funds should be used to support the Saccharomyces Genome Database project in the Department of Genetics, Stanford University. Account : GHJKO, Genetics : WAZC.”
Thank you for your support!
Kindly send by mail to:
Development Services
PO Box 20466
Stanford, CA 94309
CONTACT US:
sgd-helpdesk@lists.stanford.edu
Categories: Announcements
SGD Newsletter, December 2024
December 13, 2024
About this newsletter:
This is the December 2024 issue of the SGD newsletter. The goal of this newsletter is to inform SGD users about new features from SGD and to foster communication within the yeast community.
Contents
- 1 AlphaFold protein structures now on SGD protein pages
- 2 YeastMine data available in AllianceMine
- 3 Help maintain your genomic databases! Sign open letter supporting funding for biodata resources
- 4 Community wiki shutting down
- 5 SGD’s new GENETICS publication
- 6 microPublications – Latest yeast papers
- 7 Alliance of Genome Resources – Latest Release 7.4
- 8 Give a Gift / Support SGD
- 9 Upcoming conferences and courses
- 10 Happy Holidays from SGD!
AlphaFold protein structures now on SGD protein pages
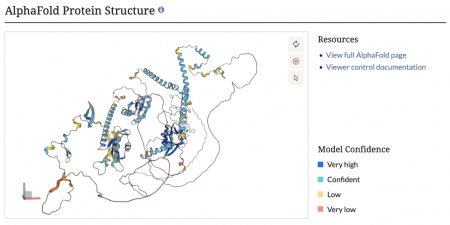
We are thrilled to announce that we have now integrated AlphaFold protein structures into our protein pages! This cutting-edge addition provides detailed, high-accuracy 3D models of protein structures, offering invaluable insights into protein function and interactions. Researchers can now explore these comprehensive structural predictions directly within SGD, facilitating advanced studies in molecular biology and bioinformatics. Dive into the new AlphaFold protein structures and elevate your research with this powerful tool!
AlphaFold, developed by DeepMind, is an AI program that accurately predicts protein structures from amino acid sequences, enabling visualization of protein conformations.
The predicted structures can be accessed through the Protein Data Bank (PDB) and AlphaFold Protein Structure Database. Thanks to Kim Rutherford and Val Wood of Pombase for tips about adding AlphaFold structures to SGD.
YeastMine data available in AllianceMine

Here at SGD we provide high-quality curated genomic, genetic, and molecular information on the genes and gene products of the budding yeast Saccharomyces cerevisiae. In 2011, SGD implemented InterMine, an open-source data warehouse system with a sophisticated querying interface, to better meet the complex and diverse needs of researchers searching and comparing data, resulting in the creation of YeastMine.
YeastMine is a multifaceted search and retrieval environment that provides access to diverse data types. Searches can be initiated with a list of genes, a list of Gene Ontology terms, or lists of many other data types. The results from queries can be combined for further analysis and saved or downloaded in customizable file formats. Queries themselves can be customized by modifying predefined templates or by creating a new template to access a combination of specific data types.
In July 2024, YeastMine was discontinued due to ongoing cuts in funding at SGD. However, we have moved the YeastMine data into AllianceMine, hosted by the Alliance of Genome Resources, of which SGD is a founding member. You can even access lists like “ALL_Verified_Uncharacterized_Dubious_ORFs” through the AllianceMine Lists just as you could in the previous versions of YeastMine. Ensure any bookmarks to YeastMine have been updated to match the new URL: https://www.alliancegenome.org/bluegenes/alliancemine. User documentation for the new YeastMine interface is available from InterMine.
Help maintain your genomic databases! Sign open letter supporting funding for biodata resources

The Global Biodata Coalition has an open letter campaign to show support for sustainable funding for biodata resources.
Please take a minute to read and sign- the form takes only seconds to fill out. Everyone – including students and postdocs, bioinformaticians and curators, PIs and directors – is invited to join those of us that have already added our signatures in support of the GBC. Find more information and sign at the GBC site.
Community wiki shutting down
As another casualty of decreased funding, SGD will no longer provide the Community Wiki. Some of the resources hosted on the wiki have been moved to our Help pages, the Alliance Community Forum, or remain available though the SGD downloads site. We thank all our past and current Community Wiki contributors, and invite the yeast community to join SGD on the Alliance Community forum.
SGD’s new GENETICS publication

Saccharomyces Genome Database: Advances in Genome Annotation, Expanded Biochemical Pathways, and Other Key Enhancements has now been published in GENETICS and is available as an accepted manuscript. Check out the most recent updates at SGD, including the two most recent reference genome annotation updates, expanded biochemical pathways representation, changes to SGD search and data files, and other enhancements to the SGD website and user interface.
microPublications – Latest yeast papers
microPublication Biology is part of the emerging genre of rapidly-published research communications. microPublications publishes brief, novel findings, negative and/or reproduced results, and results which may initially lack a broader scientific narrative. Each article is peer-reviewed, assigned a DOI, and indexed through PubMed and PubMedCentral.
Consider microPubublications when you have a result that doesn’t necessarily fit into a larger story, but will be of value to others.
Latest yeast microPublications:
- Beard JS, Francis LK, Forrest RC, Kalinowski A, Parks JC, Griffin WH, Tackett CL, Duina AA (2024) Trapping of yFACT at 3′ ends of genes is not a universal characteristic of yeast versions of Bryant-Li-Bhoj syndrome histone H3 mutants. MicroPubl Biol 2024
- Di Terlizzi M, Liberi G, Pellicioli A (2024) Separation of function mutants underline multiple roles of the Srs2 helicase/translocase in break-induced replication in Saccharomyces cerevisiae. MicroPubl Biol 2024
- Hill JM, Pedersen RT, Drubin DG (2024) Myosin-I’s motor and actin assembly activation activities are modular and separable in budding yeast clathrin-mediated endocytosis. MicroPubl Biol 2024
- Miller JM, Tragesser-Tiña ME, Turk SM, Rubenstein EM (2024) Loss of transcriptional regulator of phospholipid biosynthesis alters post-translational modification of Sec61 translocon beta subunit Sbh1 in Saccharomyces cerevisiae . MicroPubl Biol 2024
- Mucelli X, Huang LS (2024) Naming internal insertion alleles created using CRISPR in Saccharomyces cerevisiae . MicroPubl Biol 2024
- Owutey SL, Procuniar KA, Akoto E, Davis JC, Vachon RM, O’Malley LF, Schneider HO, Smaldino PJ, True JD, Kalinski AL, Rubenstein EM (2024) Endoplasmic reticulum and inner nuclear membrane ubiquitin-conjugating enzymes Ubc6 and Ubc7 confer resistance to hygromycin B in Saccharomyces cerevisiae . MicroPubl Biol 2024
- Pinto J, Tavakolian N, Li CB, Stelkens R (2024) The relationship between cell density and cell count differs among Saccharomyces yeast species. MicroPubl Biol 2024
- Ramakrishnan P, Keeney J (2024) The yeast gene ECM9 regulates cell wall maintenance and cell division in stress conditions. MicroPubl Biol 2024
All yeast microPublications can be found in SGD.
Alliance of Genome Resources – Latest Release 7.4
The Alliance of Genome Resources, a collaborative effort between SGD and other model organism databases (MODs), released version 7.4 in October 2024.
The 7.4.0 release includes ~2.5 million variants of clinical significance from ClinVar. The Alliance removed 400 million human variants from the search because their inclusion was hampering site performance. The removed variants are mostly of unknown or uncertain clinical significance. The Alliance has instead included over 2.5 million human variants of known clinical significance from ClinVar. This should speed up the performance of the search function and improve site stability. The Alliance will address inclusion of the other high throughput human variants in the future. More detailed information about the 7.4 Alliance release can be found in the release notes.
Give a Gift / Support SGD
Budget cuts from NIH continue to strain SGD’s finances. Despite our efforts at reducing costs, we still have significant ongoing budgetary challenges. Donations are now critical for our work to continue.
Your generous gift to SGD will help us to continue providing essential information for your research and teaching efforts.
To contribute, please make checks payable to Stanford University, noting that “the funds should be used to support the Saccharomyces Genome Database project, under the direction of Drs. Sherlock and Cherry in the Department of Genetics, Stanford University. Account : GHJKO, Genetics : WAZC.”
Thank you for your support!
Kindly send by mail to:
Development Services
PO Box 20466
Stanford, CA 94309
CONTACT US: sgd-helpdesk@lists.stanford.edu
Upcoming conferences and courses
- Yeast2025: 32nd International Conference on Yeast Genetics and Molecular Biology ICYGMB32
- July 21 to July 24, 2025
- Sorbonne University, Paris, France
- 38th International Specialized Symposium on Yeasts (ISSY38)
- September 01 to September 05, 2025
- Warsaw University, Warsaw, Poland
Happy Holidays from SGD!
We want to take this opportunity to wish you and your family, friends, and lab mates the best during the upcoming holidays. Stanford University will be closed for two weeks starting end of day Friday, December 20th, and reopening on Monday, January 6th, 2025. Although SGD staff members will be taking time off, the website will be up and running throughout the winter break, and we will resume responding to user requests and questions in the new year.
Note: If you wish to receive this newsletter by email, please contact the SGD Help Desk at sgd-helpdesk@lists.stanford.edu.
Categories: Newsletter
AlphaFold protein structures now on SGD protein pages
November 18, 2024
We are thrilled to announce that we have now integrated AlphaFold protein structures into our protein pages! This cutting-edge addition provides detailed, high-accuracy 3D models of protein structures, offering invaluable insights into protein function and interactions. Researchers can now explore these comprehensive structural predictions directly within SGD, facilitating advanced studies in molecular biology and bioinformatics. Dive into the new AlphaFold protein structures and elevate your research with this powerful tool!

AlphaFold, developed by DeepMind, is an AI program that accurately predicts protein structures from amino acid sequences, enabling visualization of protein conformations. The predicted structures can be accessed through the Protein Data Bank (PDB) and AlphaFold Protein Structure Database.
Thanks to Kim Rutherford and Val Wood of Pombase for tips about adding AlphaFold structures to SGD.
Categories: Announcements
YeastMine shutting down July 15
July 01, 2024
Due to ongoing cuts to our funding, SGD can no longer continue to provide the YeastMine data warehouse resource. It is with heavy hearts that we discontinue this service.
Back in 2011, SGD implemented InterMine (http://www.InterMine.org), an open source data warehouse system with a sophisticated querying interface, to create YeastMine, a multifaceted search and retrieval environment that provided access to diverse data types. YeastMine served as a powerful search interface, a discovery tool, a curation aid, and a complex database presentation format.
YeastMine has served us all quite well. We are working to move the YeastMine data into AllianceMine, hosted by the Alliance of Genome Resources, of which SGD is a founding member.
To get started with AllianceMine, go to the Templates page, and filter by category = ‘YeastMine’.
Categories: Announcements
SGD Newsletter, Summer 2024
June 20, 2024
About this newsletter:
This is the Summer 2024 issue of the SGD newsletter. The goal of this newsletter is to inform our users about new features in SGD and to foster communication within the yeast community. You can view this newsletter as well as previous newsletters, on the SGD Community Wiki.
Contents
- Give a Gift / Support SGD
- Reference genome update R64.5
- Extended gene coordinates in GFF
- Updates to SGD search
- microPublications – latest yeast papers
- Alliance of Genome Resources – Latest Release 7.2
- Upcoming conferences and courses
Give a Gift / Support SGD
Budget cuts from NIH continue to strain SGD’s finances. Despite our efforts at reducing costs, we still have significant ongoing budgetary challenges. Donations are now critical for our work to continue.
Your generous gift to SGD will help us to continue providing essential information for your research and teaching efforts.
To contribute, please make checks payable to Stanford University, noting that “the funds should be used to support the Saccharomyces Genome Database project, under the direction of Drs. Sherlock and Cherry in the Department of Genetics, Stanford University. Account : GHJKO, Genetics : WAZC.”
Thank you for your support!
Kindly send by mail to:
Development Services
PO Box 20466
Stanford, CA 94309
CONTACT US: sgd-helpdesk@lists.stanford.edu
Reference genome update R64.5

The S. cerevisiae strain S288C reference genome annotation has been updated to include previously unannotated features. The new genome annotation is release R64.5.1, dated 2024-05-29. Note that the underlying genome sequence itself was not altered; the chromosome sequences remain stable and unchanged.
The R64.5.1 update included:
- Six new open reading frames (ORFs): YDL204W-A, YFR035W-A, YGR016C-A, YMR106W-A, YNL040C-A, YNL155C-A
- New uORFs for 4 ORFs: ATG12/YBR217W, ATG19/YOL082W, ATG5/YPL149W, ATG13/YPR185W
- A uORF is a small upstream open reading frame that precedes, and regulates downstream translation of, the major ORF.
- Move start downstream: EFM4/YIL064W
- ORF upgraded from Dubious to Verified: YIL059C
Various sequence and annotation files are available on SGD’s Downloads site. You can find more update details on the Details of 2024 Reference Genome Annotation Update R64.5 SGD Wiki page.
Extended gene coordinates in GFF
The saccharomyces_cerevisiae.gff contains sequence features of Saccharomyces cerevisiae and related information such as Locus descriptions and GO annotations. The saccharomyces_cerevisiae.gff is fully compatible with Generic Feature Format Version 3, and is updated weekly.
In recent years, SGD has made two significant changes to the GFF content (described in more detail below):
- In November 2020, SGD updated the file to reflect experimentally determined transcripts
- In February 2024, SGD edited the ‘gene’ entries in the file to extend the coordinates to encompass the start and stop coordinates of the longest experimentally determined transcripts
In November 2020, SGD updated the transcripts in the GFF file to reflect the experimentally determined transcripts (Pelechano et al. 2013, Ng et al. 2020), when possible. The longest transcripts were determined for two different growth media – galactose and dextrose. When available, experimentally determined transcripts for one or both conditions were added for a gene. When this data was absent, transcripts matching the start and stop coordinates of an open reading frame (ORF) were used.
Starting November 2020: BDH2/YAL061W with rows for longest transcripts expressed in GAL and in YPD.
{kind=link}
Then in February 2024, SGD increased the start and stop coordinates of genes to encompass the start and stop coordinates of the longest experimentally determined transcripts, regardless of condition. This change was made in order to comply with JBrowse 2, a newer and more extensible genome browser, which requires that parent features in GFF files (genes) are larger than child features (mRNA, CDS, etc) (Diesh et al., 2023).
After February 2024: BDH2/YAL061W with expanded start/stop coordinates for ‘gene’, still with rows for longest transcripts expressed in GAL, YPD.
{kind=link}

GFF is a standard format used by many groups. SGD uses the GFF file to load the reference tracks in SGD’s genome browser resource.
Updates to SGD search

SGD is jam-packed with information, with new data being added every day. It’s a lot to keep up with, and with so much info, some inevitably ends up hidden from view. To make the various data types in SGD more readily accessible, we have made various improvements to the SGD search:
- New category for datasets. Over 3700 yeast datasets are accessible. Search by reference, keyword, assay, and lab.
- New Strains subcategory for Reference search. Scroll down to ‘Associated Strains’ in the lefthand menu on the Search Results page.
- Macromolecular complexes can now be searched with aliases. Further refine by reference, subunit, function, process, and location.
- Search for alleles via their descriptions and SGDIDs. Drill down based on reference, allele type, gene, and phenotype.
- RNA products can now be searched using RNAcentral IDs.
microPublications – latest yeast papers
microPublication Biology is part of the emerging genre of rapidly-published research communications. microPublications publishes brief, novel findings, negative and/or reproduced results, and results which may initially lack a broader scientific narrative. Each article is peer-reviewed, assigned a DOI, and indexed through PubMed and PubMedCentral.
Consider microPubublications when you have a result that doesn’t necessarily fit into a larger story, but will be of value to others.
Latest yeast microPublications:
- Caligaris M, De Virgilio C (2024) Proxies introduce bias in decoding TORC1 activity. MicroPubl Biol 2024.
- Fromont-Racine M, Khanna V, Jacquier A, Badis G (2024) YLR419W is the homolog of the mammalian translation initiation factor DHX29. MicroPubl Biol 2024.
- Greenlaw A, Dell R, Tsukiyama T (2024) Initial acidic media promotes quiescence entry in Saccharomyces cerevisiae. MicroPubl Biol 2024.
- Harmer ZP, Hohener TC, Landolt AE, Mitchell C, McClean M (2024) Enhancing high-throughput optogenetics: Integration of LITOS with Lustro enables simultaneous light stimulation and shaking. MicroPubl Biol 2024.
- Karpel JE (2024) Caenorhabditis elegans ddx-15 helicase fails to complement loss of Prp43p in Saccharomyces cerevisiae. MicroPubl Biol 2024.
- Medina-Suarez S, Machin F (2024) The CRISPR/Cas9 system forms a condensate in the yeast nucleus. MicroPubl Biol 2024.
- Putnam CD (2024) Loss of mitochondrial DNA is associated with reduced DNA content variability in Saccharomyces cerevisiae. MicroPubl Biol 2024.
- Rosenbaum JC, Carlson AE (2024) The SARS coronavirus accessory protein ORF3a rescues potassium conductance in yeast. MicroPubl Biol 2024.
All yeast microPublications can be found in SGD.
Alliance of Genome Resources – Latest Release 7.2
The Alliance of Genome Resources, a collaborative effort between SGD and other model organism databases (MODs), released version 7.2 in June 2024.
The 7.2.0 release updates the Associated Alleles and Associated Models tables on Disease pages:
- Each table has a new column, Disease Qualifier, with a working filter. The qualifier describes whether an allele or model may be, for example, implicated in the onset of a disease or a model for the severity of a disease, respectively
- In addition to the Disease Qualifier, the Associated Models table now has new columns for Condition Modifier and Genetic Modifier
- The “Annotation Details” pop-up has expanded to include more information.
- Alleles table: Association, Genetic Modifiers, Genetic Sex, Notes, and Annotation Type
- Models table: Genetic Sex, Notes, and Annotation Type
- The Associated Models table now has working filters for the Experimental Condition, Condition Modifier, and Genetic Modifier columns, including the ability to filter on relationship (e.g. induced by) as well as content (e.g. “copper”)
- The Download files from the disease page Associated Alleles table and Associated Models table now include additional information as well.
- New columns and information for the Associated Alleles table include: Allele Association, Genetic Entity Association, Disease Qualifier, Evidence Code Abbreviation, Experimental Conditions, Genetic Modifier Relation, Genetic Modifier IDs, Genetic Modifier Names, Genetic Sex, Notes, Annotation Type, Source URL, and Date.
- New columns and information for the Associated Models table include: Model Type, Model Association, Disease Qualifier, Evidence Code Abbreviation, Experimental Conditions, Condition Modifiers, Genetic Modifier Relation, Genetic Modifier IDs, Genetic Modifier Names, Genetic Sex, Notes, Annotation Type, Source URL, and Date.
Upcoming conferences and courses
- RCN-UBE: Yeast ORFan Gene Project – Summer Workshop – Gene Expression Analysis
- June 20 to June 21, 2024
- Virtual
- FASEB Yeast Chromosome Biology and Cell Cycle
- June 23 to June 27, 2024
- Fort Garry Hotel, Winnipeg, Manitoba, Canada
- JCS2024: Diversity and Evolution in Cell Biology
- June 24 to June 27, 2024
- Montanya Hotel & Lodge, Catalonia, Spain
- Yeast Genetics & Genomics
- July 23 to August 13, 2024
- Cold Spring Harbor Laboratory, Cold Spring Harbor, New York
- Pacific Northwest Yeast Club
- July 26, 2024
- Fred Hutchinson Cancer Center, Seattle, WA
- 39th Small Meeting of Yeast Transporters and Energetics (SMYTE)
- August 28 to September 01, 2024
- University of York, York, United Kingdom
- ICY2024: 16th International Congress on Yeasts
- September 29 to October 03, 2024
- Cape Town International Convention Centre, Cape Town, South Africa
- Yeast2025: 32nd International Conference on Yeast Genetics and Molecular Biology ICYGMB32
- July 21 to July 24, 2025
- Sorbonne University, Paris, France
Categories: Newsletter